导读:
近期,墨尔本大学研究团队首次提出了一种无模板微流控制备水包水微胶囊的新策略,突破了传统微胶囊制备依赖不混溶两相体系的限制,实现了在完全互溶的水单相体系(AOPS)中通过局部凝聚形成微胶囊。相关研究以“Template-Free Microfluidic Fabrication of Water-in-Water Microcapsules for Controlled Release”为题目,发表于期刊《ACS Applied Materials & Interfaces》。
本文要点:
1、研究以带正电的壳聚糖为内相、带负电的表面活性剂(SLES+CAPB)为外相,在3D打印微流控芯片中通过脉冲式外相流控制两相混合边界的局部复合凝聚,形成连续的胶囊壳层,无需外相模板或后续核材去除步骤。
2、该方法制备的微胶囊直径达数百微米,形貌均一,在富表面活性剂环境中可稳定存在超4个月且无聚集、崩解,壳层形成具有可逆性——在碱性环境中快速溶解,酸性及特定离子环境下保持完整。
3、实验表征显示,微胶囊壳层厚度约218±74 nm,杨氏模量为20-30 MPa,兼具机械柔性与稳定性;其壳层具有尺寸选择性渗透性,可有效截留500 nm及以上的颗粒,而小分子溶质能自由扩散,静电作用还会影响包封颗粒在胶囊内的分布。
4、此外,微流控的脉冲参数是调控胶囊形貌的关键,优化后可制备近球形微胶囊,且该方法材料灵活性高、环境友好,具备规模化潜力。
5、该研究为水单相体系中微胶囊的制备提供了新途径,克服了传统方法需有机溶剂/油相的弊端,拓宽了可持续微胶囊设计的可能性,在食品、制药、个人护理等领域的水基配方控释应用中具有重要前景,同时也为后续调控胶囊尺寸、探索外刺激响应性及工业规模化奠定了基础。
本研究制备水包水微胶囊的核心创新点是什么?与传统方法相比有哪些优势?
核心创新点为首次在完全互溶的水相单相体系(AOPS)中实现无模板微流控制备水包水微胶囊,通过控制带相反电荷的聚合物-表面活性剂流束在混合边界诱导局部凝聚形成壳层,无需依赖不互溶两相、预形成界面与后续脱核/脱模板步骤。
与传统方法相比,优势体现在:①绿色环保,无需有机溶剂/油相,适用于全水相制剂;②材料灵活,基于聚合物-表面活性剂凝聚,可通过成分调控实现性能定制;③制备精准,微流控结合脉冲流可调控胶囊形貌与尺寸,粒径均一;④工艺简单,潜在可规模化,且胶囊兼具稳定性、响应性与尺寸选择性,应用场景更广。
微流控系统的设计与操作参数是实现AOPS中无模板微胶囊制备的关键,具体作用如下:
① 3D打印芯片的针状结构:减少胶囊与通道壁的接触,避免液滴破裂,营造3D形成环境,为局部凝聚提供稳定的反应位点;
②对称稀释通道:调控外相表面活性剂浓度,增强壳层完整性,同时产生背压效应、抑制惯性迁移,减少胶囊的伸长与变形,优化形貌;
③脉冲外相流操作(1s通/10s停):解决了互溶水相无法形成稳定界面、难以实现液滴脱离的问题,暂停阶段让凝聚层在针端形成并稳定,通流阶段实现胶囊的脱离与输送,且脉冲参数可精准调控胶囊形貌;
④流束速率控制:内相恒流保证聚合物供应稳定,外相脉冲流速与稀释相流速配合,控制混合边界的浓度梯度,从而调控局部凝聚的速率与壳层形成效果,确保胶囊结构完整。
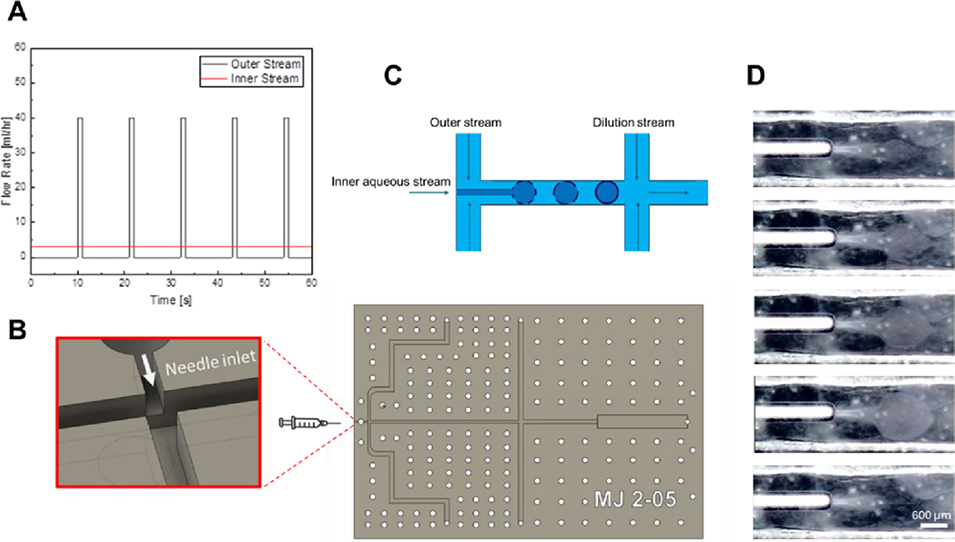
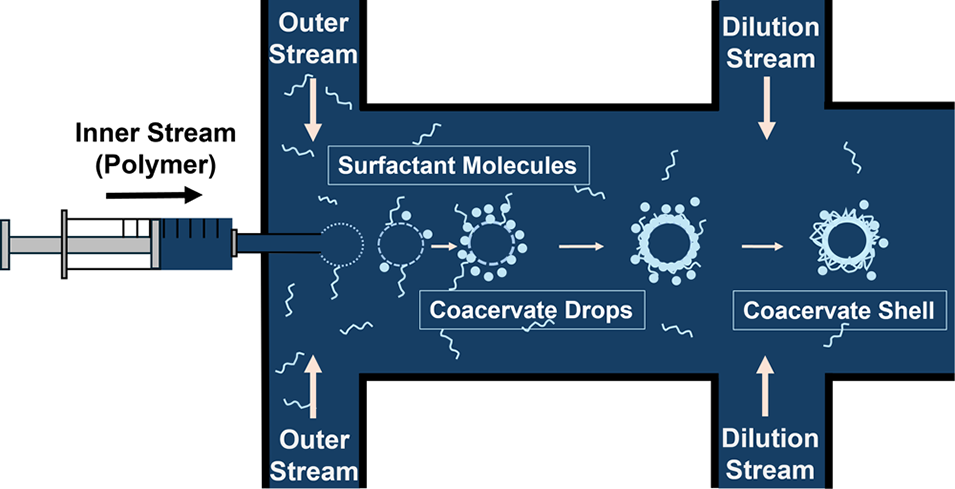
图1.(A)外水相流(9%脂肪醇聚氧乙烯醚硫酸钠、1%椰油酰胺丙基甜菜碱、1%氯化钠)的流速设置,脉冲流以40mL/h的流速持续1秒后停止10秒(脉冲时间可调整),循环往复;内水相流(0.1%壳聚糖、0.9%聚环氧乙烷、1%乙酸)以3mL/h或更高流速持续泵入。(B)微流控器件示意图,含针头适配结构。(C)微流控通道内微胶囊的形成示意图。(D)内相聚合物流通过针头缓慢注入微流控通道,此时表面活性剂流暂停,聚合物溶液形成准球形并稳定在针头尖端附近;在该静置阶段,聚合物-表面活性剂薄膜开始在聚合物液滴周围形成;当外相表面活性剂流恢复时,微胶囊脱离针头并向前推进。

图2. Hirox光学显微镜下微胶囊的典型形貌:(a)准球形微胶囊(长径990μm、短径891μm,形状指数为0),由脉冲时长1秒、脉冲间隔10秒的条件制备而成;(b)短尾型微胶囊(长径974μm、短径741μm,形状指数为1.55),由脉冲时长0.75秒、脉冲间隔7秒的条件制备而成;(c)长尾型微胶囊(长径476μm、短径436μm,形状指数为4.4),由脉冲时长0.75秒、脉冲间隔3秒的条件制备而成。
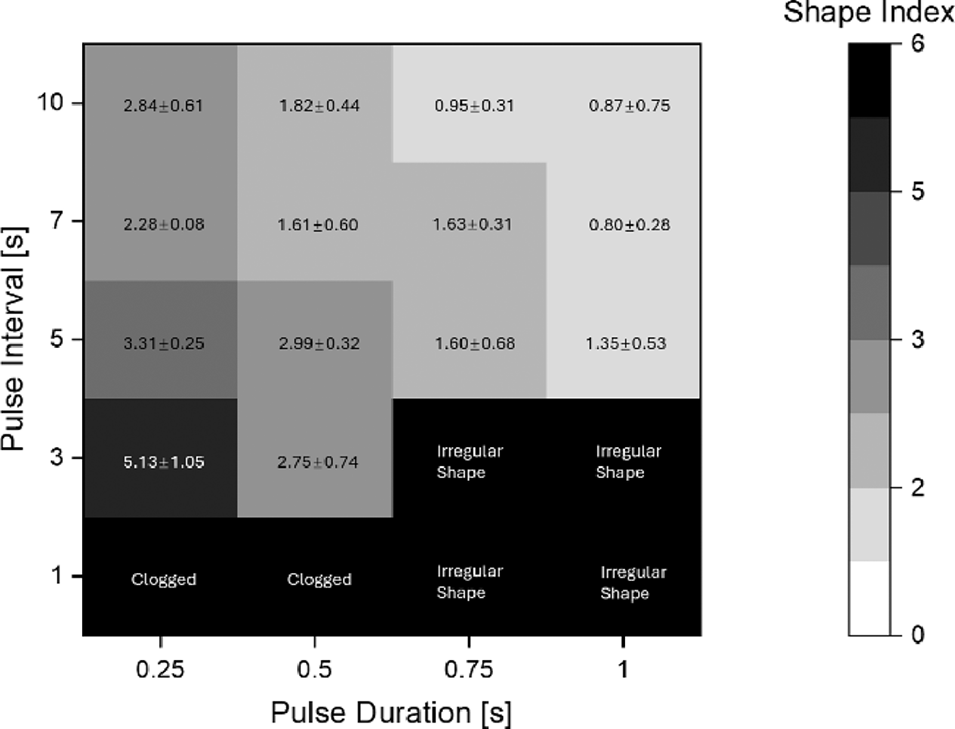
图3. 微胶囊的存在状态及形貌分布图,以形状指数(尾长与微胶囊平均粒径的比值)表征,横坐标为外相流脉冲时长,纵坐标为脉冲间隔(所有其他参数保持不变)。所有微胶囊均通过广域光学显微镜测量,长度单位为微米;浅色轮廓代表形状指数更小,对应形貌更接近球形的微胶囊;深色区域代表形貌不规则的蠕虫状微胶囊,或出现内相流堵塞、无法形成完整微胶囊的情况。
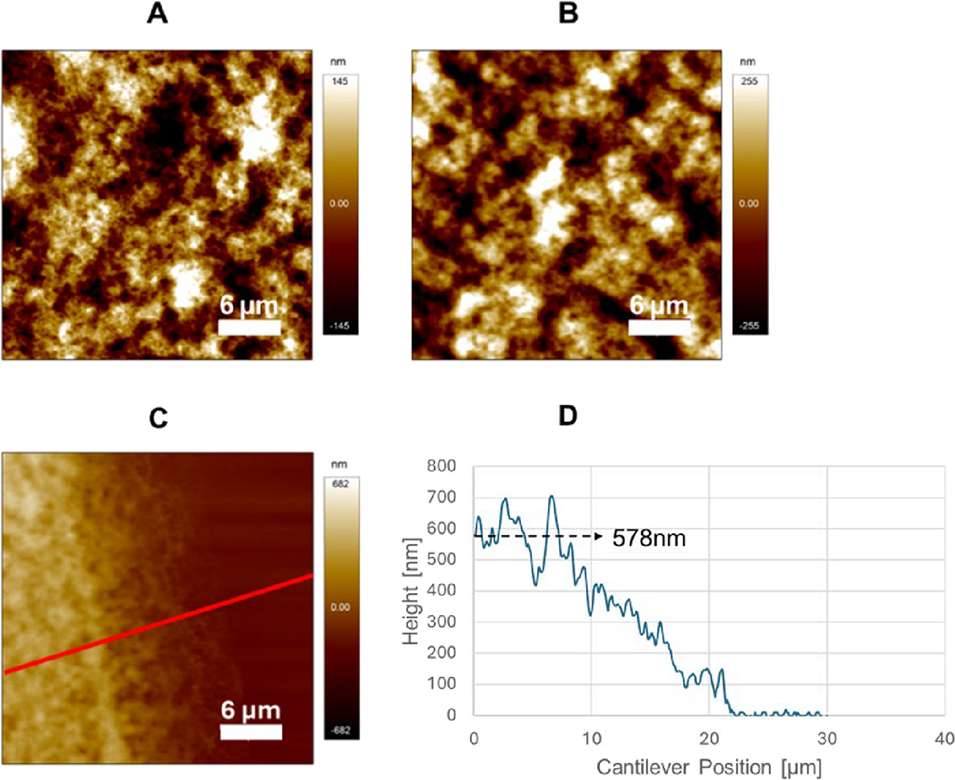
图4. 微胶囊干燥前的原子力显微镜形貌图:(A)空气中的微胶囊表面形貌;(B)浸泡在1%储存液中的微胶囊表面形貌;(C、D)空气中微胶囊边缘的原子力显微镜形貌及高度追踪(沿红色高亮线)。虚线箭头指示塌陷囊壳边缘的平均壳层高度,基底水平设为0,据此估算出壳层厚度约为289nm(为平均高度的一半)。

图5. 玻璃盖玻片上干燥后子弹形微胶囊的扫描电镜与原子力显微镜图像,样品在成像前经超纯水冲洗以去除残留表面活性剂和盐类:(A、B)不同放大倍数下微胶囊表面的二次电子图像,表面的凹凸不平可能源于胶囊制备过程中形成的凝聚层网络,或清洗过程中的部分溶胀/溶解;(C)背散射电子图像,显示微胶囊表面存在细微的成分差异,浅灰色区域表明原子序数更高的分子更易在此参与凝聚并发生后续沉淀;(D)同一样品的原子力显微镜形貌图。

图6. 微胶囊的储存稳定性及壳层在外界作用下的逐步解体过程(微胶囊储存于含0.9%脂肪醇聚氧乙烯醚硫酸钠、0.1%椰油酰胺丙基甜菜碱、1%氯化钠的培养皿中):(A)从微流控器件中新制备的微胶囊;(B、C)在体系中加入1mL 5%氢氧化钠溶液后,不同时间点通过Hirox光学显微镜观察到的微胶囊结构变化;(D)同批次微胶囊在室温下储存4个月后的状态;(E)储存4个月的微胶囊经盖玻片手动挤压、揉搓后的形态变化(E1为挤压前,E2为挤压后,E3为揉搓后)。

图7. Hirox光学显微镜拍摄的包封不同粒径聚苯乙烯微球的微胶囊光学显微照片:(A)5μm聚苯乙烯微球;(B)1μm聚苯乙烯微球;(C)800nm聚苯乙烯微球;(D)500nm聚苯乙烯微球。
论文链接:https://doi.org/10.1021/acsami.6c00242
(本文仅供参考学习及传递微流控研究成果,版权归原作者所有,如侵犯权益,请联系删除)